Within each of our cells, two meters of DNA are folded into a nucleus of 10 microns diameter. The integrity and functionality of the genome, which are essential for cell survival, particularly through the adequate regulation of transcription, are preserved by histones. These proteins associate to form the nucleosome, the basic unit of chromatin. This structure is repeated and organized at different scales, leading to the most compact structure constituted by the metaphase chromosome. While sequencing a genome has become a routine task, characterizing histones is still an analytical challenge because of subtle variations in amino acid sequences between isoforms and histone variants, and because of the many covalent modifications that decorate them in a dynamic way.
To date, more than 80 histone sequences have been described, encoded by different genes and / or from alternative splicing events. Some histones have very different sequences, for example due to the addition of an entire macromolecular domain. Other histone sequences vary by only a few amino acids distributed within the protein sequence. The description of all histone forms has been made over the years within different species.
To contribute to the collective effort to define a precise and unambiguous nomenclature for histones, and to provide a list of reference sequences useful for proteomic analyses of these proteins, the
team “Exploring the Dynamics of Proteomes” of the Large-Scale Biology laboratory have previously performed a patient inventory of histone sequences present in human and mouse
[1]. This work has provided a solid foundation for the development of a targeted proteomic method allowing the quantification of a maximum number of histone variants of H2A and H2B
[2].
From an extract of chromatin-linked proteins consisting of approximately 1500 proteins, the researchers were able to identify 25 variants, including sequences varying by a subtle "teasing game" between amino acids. They were able to demonstrate an increased abundance of several isoforms of H2A.L.1 at the end of mouse spermatogenesis and to reveal a higher abundance of these isoforms in a mouse model of male infertility.
This protein sequence database will also be very useful to explore the multitude of histone post-translational modifications that contribute to finely regulating chromatin structure and function
[3].
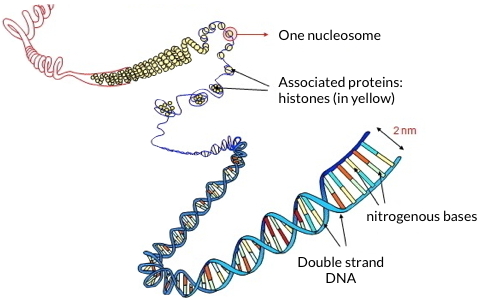
Schematic representation of DNA folding, showing associated histones to form the nucleosome.