En science expérimentale, la collecte d’observations peut être affectée par des valeurs manquantes (définies par une absence de mesures pour une observation donnée). Un trop grand nombre de valeurs manquantes pouvant compromettre l’analyse, l’imputation (c’est-à-dire la complétion des données par l’estimation des mesures qui auraient dues être observées) est dans bien des cas une nécessité et un moindre mal. Cependant, cela est particulièrement difficile en
protéomique*, en raison du nombre de valeurs manquantes, mais aussi de leurs multiples origines.
Des chercheurs du
CEA-Irig/BGE ont donc proposé un nouveau modèle statistique décrivant conjointement deux types de valeurs manquantes : celles censurées (quand un fragment de protéine n’est pas suffisamment abondant pour être détecté), et celles manquant aléatoirement (résultant de la non-exhaustivité des instruments). De plus, ils ont montré qu’à partir de ce modèle il est possible de dériver un algorithme d’imputation qui maximise les corrélations connues entre biomolécules (protéines et leurs fragments, transcrits, etc.). Enfin, en l’absence de solution analytique à ce problème de maximisation, ils ont implémenté une résolution numérique par réseau de neurones à propagation directe.
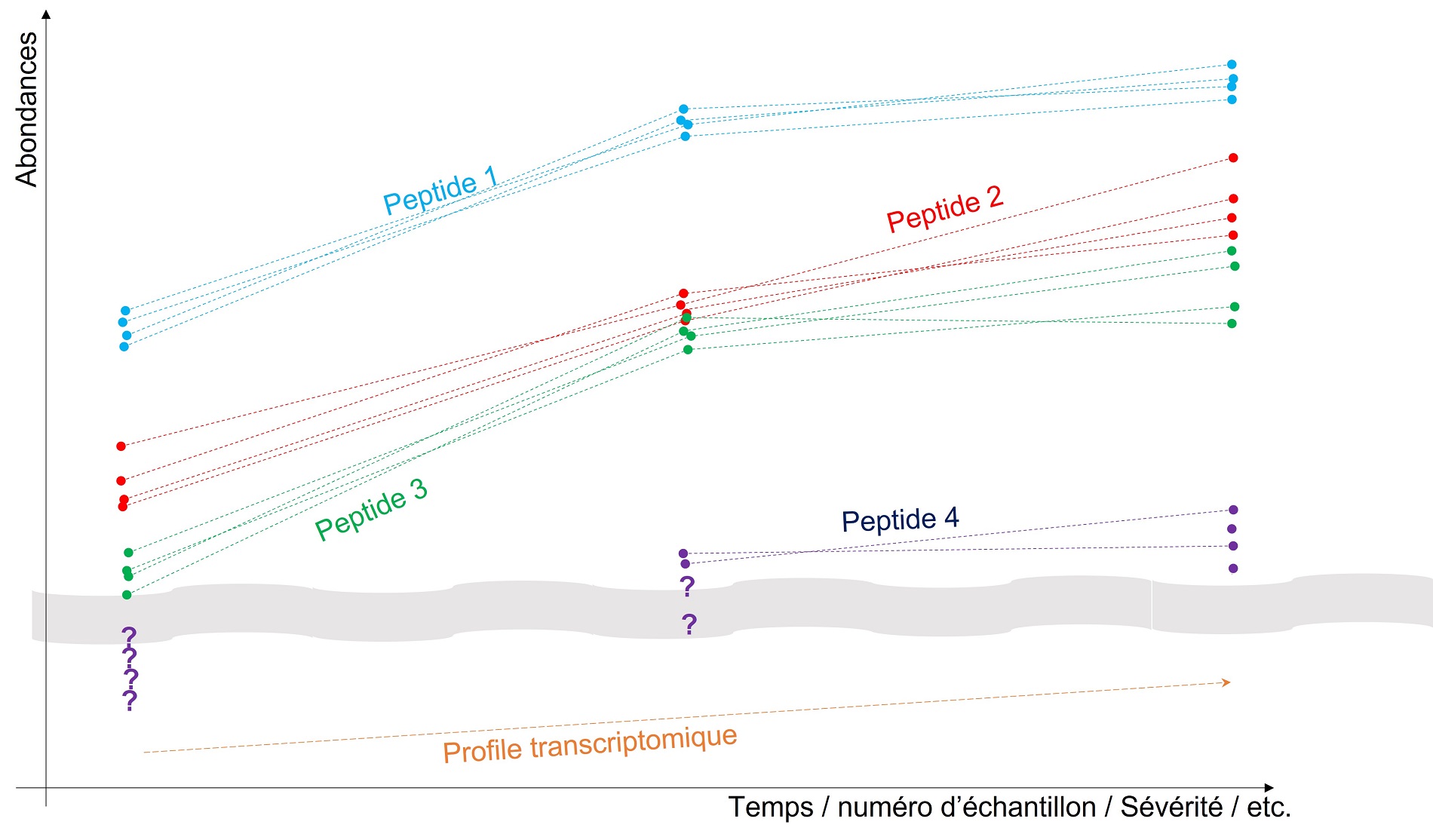
Figure : Illustration schématique de l’usage des corrélations entre différentes biomolécules pour réaliser une imputation de valeurs manquantes : plusieurs peptides provenant d’une même protéine (ainsi qu’éventuellement le transcrit ayant permis sa traduction) devant avoir des profils de mesures corrélés, il est pertinent d’imputer les valeurs manquantes afin de maximiser cette corrélation, comme illustré par l’emplacement des « ? » pour le Peptide 4.
L’outil d’imputation ainsi obtenu surpasse toutes les méthodes d’imputation de l’état de l’art, et son utilisation permet d’améliorer significativement les résultats des analyses protéomiques par spectrométrie de masse.
Protéomique* : caractérisation par identification et quantification de l’ensemble des protéines présentes dans un échantillon biologique.
FinancementsTravail soutenu par l’ANR via les projets suivants :
- ProFI (ANR-10-INBS-08)
- GRAL CBH (ANR-17-EURE-0003)
- SECRET (ANR-22-CE45-0026)
- DEAP (ANR-15-IDEX-02)
- MIAI @ Grenoble Alpes (ANR-19-P3IA-0003)
CollaborationLaboratoire Recherche Translationnelle et Innovation en Médecine et Complexité (TIMC) Univ. Grenoble Alpes, CNRS, Grenoble INP