In experimental science, data collections can be affected by missing values (defined by an absence of measure for a given observation). As too much missing values may jeopardize the data analysis, imputation (i.e., the completion of the data by estimating the measures which should have been observed) is often both a necessity and a lesser evil. However, this task is particularly difficult in
proteomics*, because of the rate of missing values, but also because of their multiple origins.
Researchers with
CEA-IRIG/BGE have therefore designed a new statistical model, which jointly characterizes two missing types of values: the censored ones, (i.e., when a protein fragment is not abundant enough to be detected), and those lacking randomly (i.e., resulting from the non-exhaustiveness of the instruments). In addition, they have shown that an imputation algorithm which maximizes the known correlations between biomolecules (proteins and their fragments, transcribed, etc.) can be derived from this model. Finally, in the absence of a formal solution to the associated maximization problem, they have implemented a numerical solver relying on a feed-forward neural network.
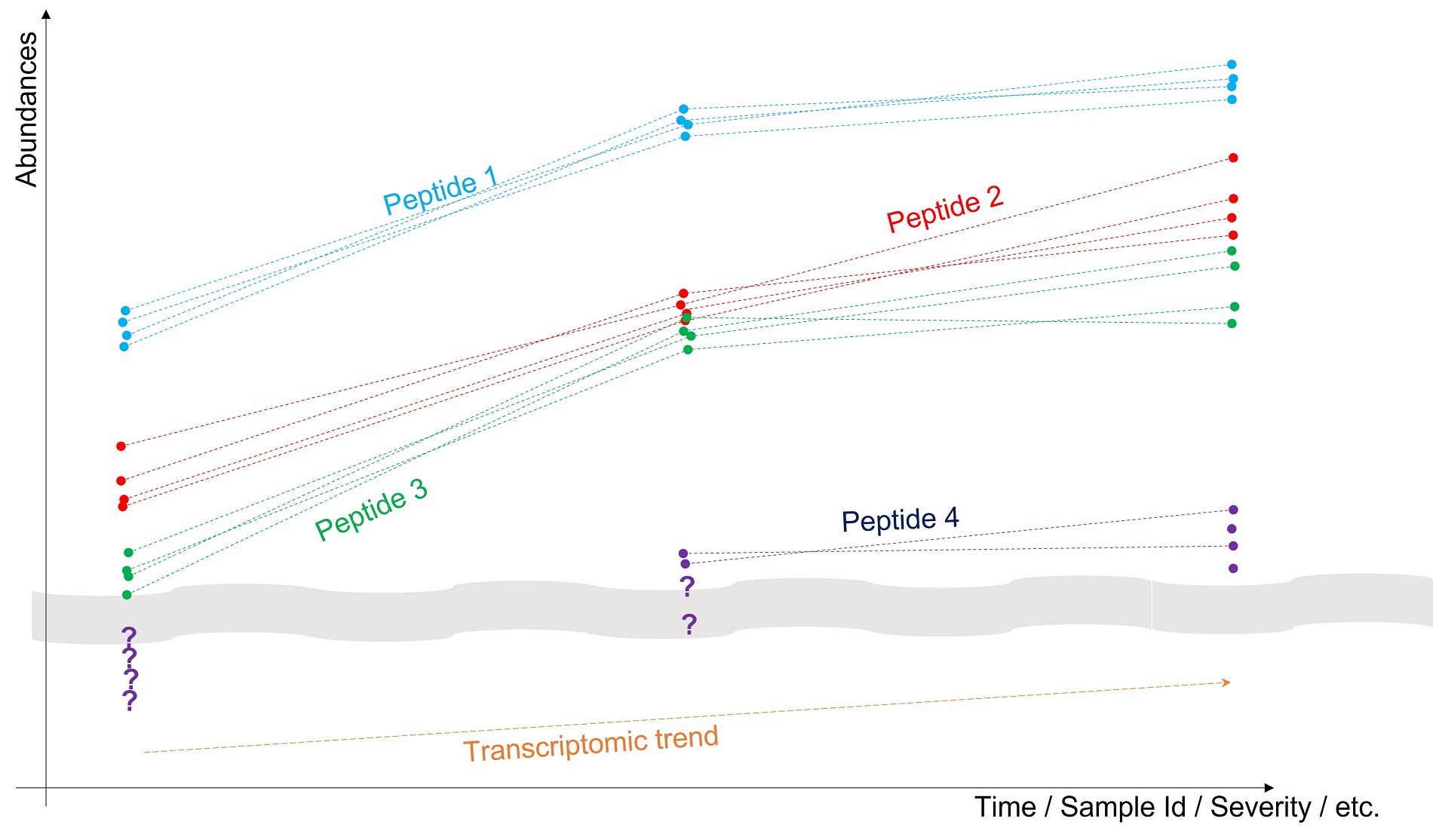
Figure: Toy example of how to leverage biomolecules’ correlations to improve missing value imputation: several peptides coming from the same protein (as well as possibly the transcript it was translated from) having measurement profiles that should be correlated. It is thus relevant to impute the missing values as to maximize it, as illustrated by the location of "?” For Peptide 4.
The resulting imputation tool outperforms all state-of-the-art imputation methods and its use makes it possible to significantly improve on the results of mass spectrometry-based proteomic analyses.
Proteomics*: characterisation by identification and quantification of all the proteins present in a biological sample.
Fundings
This work was supported by the ANR through the following projects:
- ProFI (ANR-10-INBS-08)
- GRAL CBH (ANR-17-EURE-0003)
- SECRET (ANR-22-CE45-0026)
- DEAP (ANR-15-IDEX-02)
- MIAI @ Grenoble Alpes (ANR-19-P3IA-0003).
CollaborationLaboratoire TIMC (Univ. Grenoble Alpes, CNRS, Grenoble INP) « Recherche Translationnelle et Innovation en Médecine et Complexité »