Les Baculovirus sont des virus ayant un génome circulaire à double brin d'ADN (dsDNA) qui infectent spécifiquement les cellules d'insectes, jouant un rôle important dans la régulation de leurs populations. Ils sont ainsi largement utilisés comme agents biologiques dans l'agronomie. Par ailleurs, ils sont utilisés comme système d'expression, constituant un outil biotechnologique de choix pour la production de protéines recombinantes en culture de cellules d'insectes.
Bien que couramment utilisé, les études structurales portant sur l'assemblage de la nucléocapside* des Baculovirus à l'échelle moléculaire restent limitées. Une meilleure compréhension de cette organisation permettrait de mieux appréhender son fonctionnement et notamment son efficacité en tant que système d'expression.
Dans cette étude, les chercheurs ont étudié le Baculovirus « Autographa californica multiple nucleopolyhedrovius » (AcMNPV). Ce virus est constitué d'une nucléocapside, qui est entourée d'une membrane lipidique dans laquelle des glycoprotéines virales sont insérées. La nucléocapside forme une structure allongée de 50 nm de large et d'environ 300 nm de long en moyenne dans laquelle on distingue deux parties terminales distinctes, le capuchon « apical » et la structure « basale », qui sont reliées entre elles par la capside. A l'aube de cette étude, de nombreuses protéines structurales constituant la nucléocapside n'étaient encore ni localisées, ni même identifiées.
En utilisant la cryo-ME, les chercheurs se sont particulièrement intéressés aux structures « basale » et « apicale » de la nucléocapside. Ils ont ainsi obtenu plusieurs cartes 3D allant de la haute à la moyenne résolution, permettant de pouvoir identifier et positionner les protéines constituant chacune de ces cartes 3D. La nucléocapside de AcMNPV peut être décrite par plusieurs sous-ensembles protéiques distincts ayant leur propre symétrie. En élucidant pour la première fois ces différentes symétries au sein de ces sous-ensembles, un modèle pseudo-atomique représentant toutes les parties symétriques de la nucléocapside de AcMNPV a pu être obtenu. Pour y parvenir, au niveau des meilleures résolutions des cartes 3D (inférieures à 4 Å), ils ont pu assigner des densités de nature inconnue, en utilisant « Modelangelo* ». Pour les cartes 3D de résolution moins bonne (supérieure à 4 Å), ils ont identifié et positionné plusieurs protéines à partir de structures prédites par « Alphafold* ». AlphaFold a été utilisé non seulement pour prédire les structures des protéines individuelles parmi les 155 codées par le génome de AcMNPV, mais également pour suggérer d'éventuels partenaires d'interaction, facilitant ainsi l'interprétation d'assemblages complexes.
Ces résultats ont finalement permis d'identifier huit nouvelles protéines ainsi qu'un court segment du génome viral au niveau du domaine « apical » de la nucléocapside, apportant une contribution expérimentale au rôle proposé de point d'entrée et de sortie de l'ADN viral.
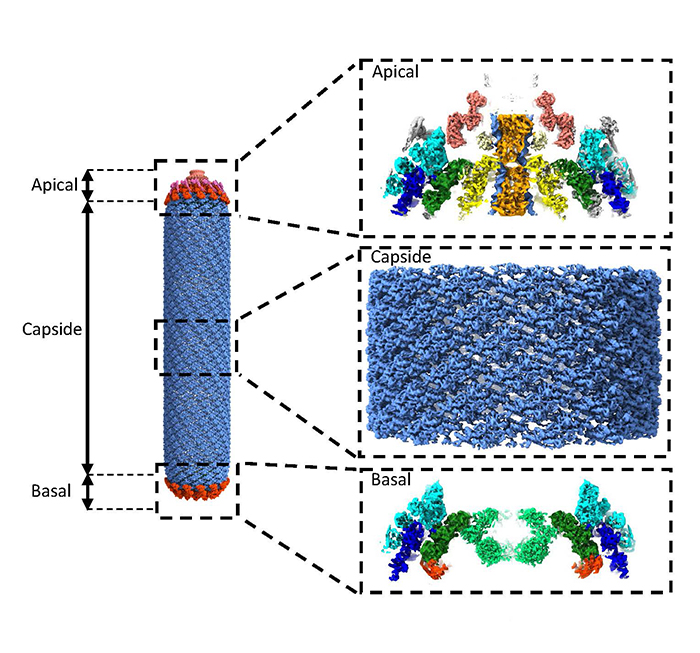
© CEA-Irig/IBS/G. Effantin
Structure
de la nucléocapside entière de AcMNPV obtenue par cryo-ME. A droite, de haut en
bas : zoom sur une vue de côté du capuchon « apical » (la
« porte d’entrée et de sortie » de l’ADN viral), la capside et une
vue de côté de la structure « basale ».
En utilisant la cryo-ME à haute résolution combinée à des algorithmes d'IA, les auteurs de cette étude ont déterminé pour la première fois la structure entière - à résolution quasi-atomique - de la nucléocapside du Baculovirus AcMNPV. Ce travail apporte une compréhension approfondie du fonctionnement des Baculovirus en enrichissant la base de données structurales du virus, et va certainement contribuer à un développement plus rationnel des outils biotechnologiques basés sur les Baculovirus.
Nucléocapside* : ensemble constitué de la capside du virus et de son acide nucléique (ADN ou ARN), le génome viral.
Modelangelo* : programme d'apprentissage automatique conçu pour construire des modèles atomiques de protéines (de séquence d'acides aminés connue ou inconnue) à partir de cartes de cryo-ME.
Alphafold* : programme d'apprentissage automatique qui prédit la structure 3D des protéines à partir de leur séquence d'acides aminés. En 2024, Alphafold offrait un accès libre à plus de 200 millions de prédictions de structures protéiques.
Collaborations :
European Synchrotron Radiation Facility (ESRF), Grenoble, France
European Molecular Biology Laboratory (EMBL), Grenoble, France