Calculating the electronic properties of organic materials is an essential key to understanding and controlling the physical mechanisms at work in organic photovoltaic (PV) or OLED (organic light-emitting diode) devices. Whether it is for the study of the optical properties of these materials, or more indirectly for the study of transport properties, via doping mechanisms in particular, IRIG researchers have for several years been developing methodologies based on
N-body perturbation theories, and more precisely the
GW theory allowing
ab initio access (
i.e. from first principles) to quantities of interest for physical systems of experimental interest.
The most widely used and digitally least
expensive method of calculating electronic structures (DFT,
Density Functional Theory) applies only to systems that are in their fundamental state. The GW method lies at the roots for calculation of molecular excitations such as absorption spectra, photo-emission spectra or fluorescence. But calculating the excitations of a 100-atom molecule by the GW method, although possible, requires the use of supercomputers.
The resulting collaboration between researchers from the IRIG and the Néel Institute has led to a series of theoretical innovations that have significantly reduced the computational complexity of the GW method. Coupled with other methods set up by these teams, their new developments pave the way for the first time to the simulation of very large systems (of the order of a thousand atoms) in a complex electrostatic environment.
These developments, implemented in the massively parallel computing code beDeft, are currently the subject of a major challenge on the
AMD Rome extension of the Irene supercomputer at the CEA's Very Large Computing Centre (TGCC) in Bruyères-le-Châtel, with the aim of demonstrating the first all-electron GW calculation on a system of a thousand atoms.
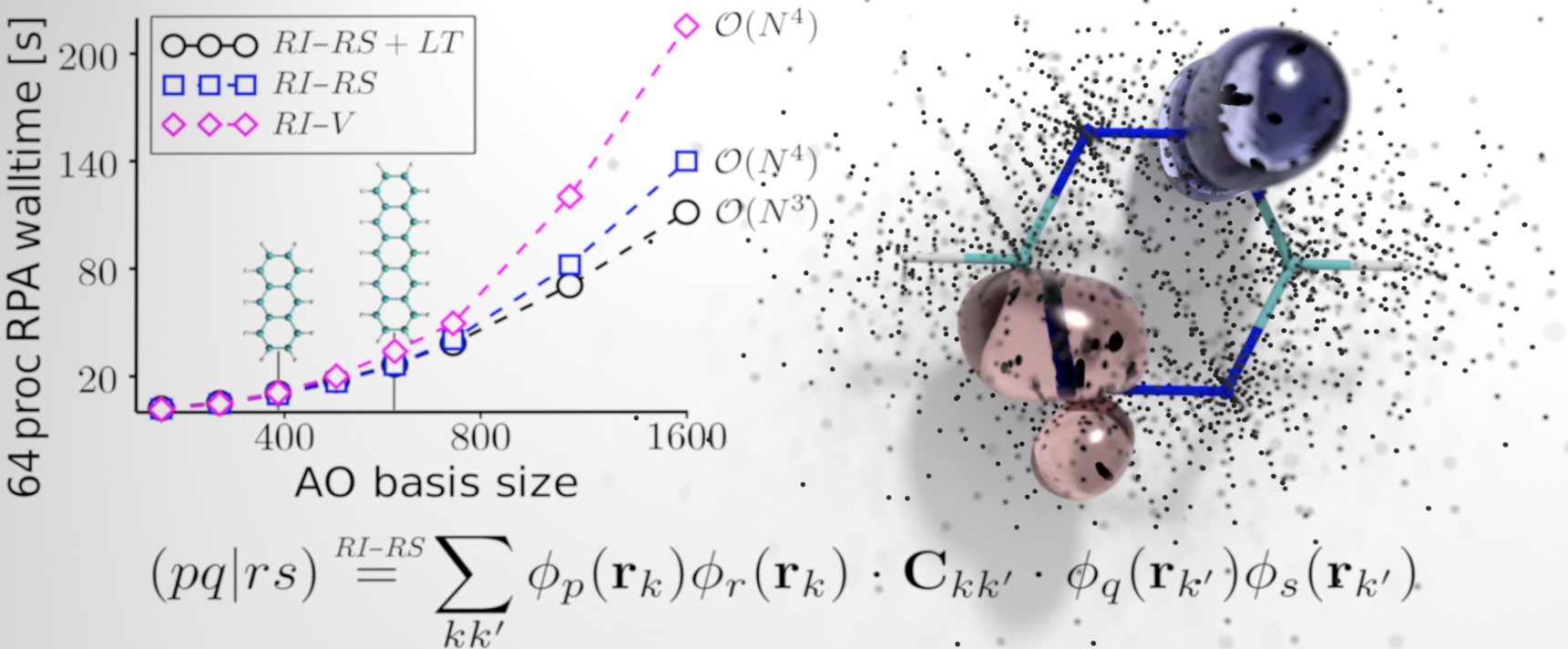
Numerical cost of different GW methods for calculating coulombic interactions as a function of the number of atomic orbits. Illustration of the GW method in real space with the point cloud considered for the benzene molecule.
This work is thus based on two fundamental innovations: the formulation of a separable identity resolution technique, based on a real space approach (illustration) on the one hand, and on the other hand the application of complex analysis techniques known as analytical continuation techniques to the description of the screened coulombic potential (W), a central ingredient of the GW methodology.
N-body perturbation theory: this theory, coming from quantum mechanics, allows to calculate the interactions between several electrons using a perturbation processing of a system without interaction. The interaction is considered as a disturbance and the different disturbance orders are summed up to correct the calculated quantities.
The
GW theory is based on Hedin's equations from the N-body perturbation theory which considers a set of solutions coupling different terms including the response function, the shielded potential (W) and the Green's system function (G).
The usual GW method has a
numerical cost varying to the power 4 of the number of atoms O(N4). If the number of atoms is multiplied by two, the calculation is multiplied by 16! The O(N3) method developed will save half of the calculation time initially required when the size of the system is doubled.
Rome is codename for AMD's high-performance server microprocessors.