Within a living organism, one cell type is differentiated from another by its own gene expression program. Genes are encoded by DNA that wraps around proteins called
histones to form a structure called chromatin. Gene expression is closely controlled by the dynamic modification of histones through the addition of covalent chemical groups on certain amino acids. The most well-known modifications are methylation and acetylation of histone lysine. Their diversity has greatly increased over the last decade with the discovery of a wide range of acylations, structures that are fairly close to acetylation but which give to modified lysines different physico-chemical properties. These acylations are added to lysines from the corresponding Acyl-CoA small molecules (metabolites). A key question that has emerged from the discovery of these new modifications, known as
epigenetic marks, is whether they provide redundant functions with acetylation or whether they have specific roles, particularly in the dynamics of chromatin structure (open or compacted) and the control of gene expression.
Among acylations, crotonylation is a modification of lysine residues which consists of the addition of a crotonyl group whose particularity is its rigid planar structure. To better understand the role of this epigenetic mark ignored until 2011, researchers at our institute have analyzed its dynamics during the process of cellular differentiation represented by murine spermatogenesis. By proteomic analysis, they observed in particular that the crotonylation of lysine 27 on histone H3 (H3K27cr) was similar in abundance to the acetylation of the same amino acid (H3K27ac). Lysine acetylation on histones has been explored for more than 50 years, and H3K27ac is known to mark the active expression of genes when present upstream of their sequence. Given the substantial abundance of H3K27cr, it seemed very opportune to finally highlight its cellular functions in relation to those of H3K27ac. The researchers then obtained the genomic location of this new histone mark and compared it to that of H3K27ac, as well as to that of proteins involved in the regulation of gene transcription. They observed that these two marks often functioned synergistically, inducing maximal gene expression. However, some regions of the genome carry more one mark than the other, which results in preferentially attracting different panels of transcriptional regulators.
This integrated analysis of omics data provides an unprecedented level of understanding of the regulation of gene expression of these two histone marks and reveals the synergistic and specific actions of each histone modification. Knowledge of all modified histone forms, as well as the proteins that bind to them, is essential for understanding the molecular mechanisms regulating gene transcription at work in normal developmental processes, but also in many pathologies.
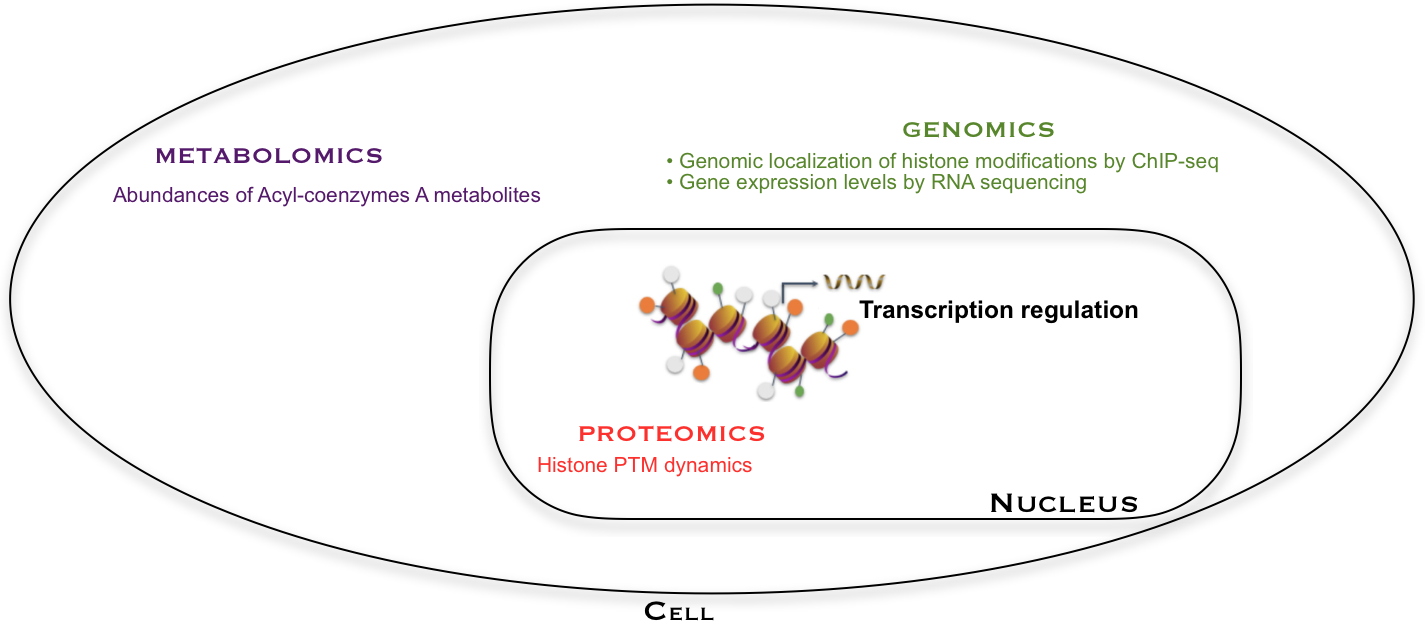
Gene expression is analyzed by RNA-seq (a sequencing technique). It is regulated by histone modifications, in particular on lysines whose position on the histone sequence and level of modification can be apprehended by proteomic analysis. Lysine acylations are introduced from Acyl-CoA co-factors, whose abundances are quantified by metabolomic analysis. The position of a histone mark on the genome (such as H3K27cr) is obtained by another type of genomic analysis (ChIP-seq, a technique for studying DNA/protein interactions at the genome level). These various analyses have been combined in this work to better understand the mechanisms regulating gene expression.
Histones are proteins located in the nucleus of eukaryotic cells. They are the main protein constituents of chromosomes and are closely associated with the DNA, and allow their compaction. The DNA is in fact wrapped around the histones like thread around a coil. Histones are very rich in the basic amino acids lysine and arginine.
Epigenetic marks are placed on the genome and are used to select the genes to be expressed. These are "post-it" notes that mark the RNA biomolecules to be synthesized.
Collaboration Institut Cochin and CEA-Saclay