Immunotherapies have become a reference treatment for advanced cancers, leading to improved overall survival. Nevertheless, only a limited proportion of patients benefit, primarily because reliable molecular biomarkers for identifying responders are still lacking. Here, we introduce an
in silico approach based on gene co-expression network analysis to derive predictive signatures of immunotherapy response in renal cancer patients.
We constructed gene co-expression networks and analyzed their topological properties to assess their utility in predicting therapeutic response in renal cancer patients. Based on a cohort of over 300 renal tumor transcriptomes, we generated patient-specific gene networks (Figure, panel A) and evaluated their connectivity, gene-gene associations, network similarity, and pathway deregulation (Figure, panel B). This analysis uncovered gene co-expression signatures and enhanced the predictive performance of machine learning models for immunotherapy response (Figure, panel C).
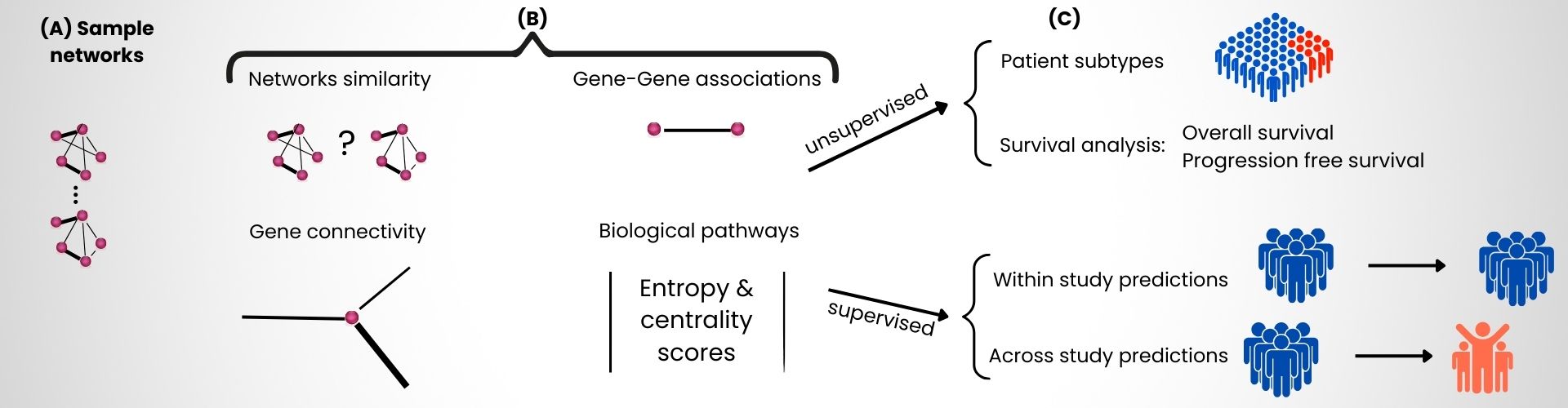
Figure: Overview of the in silico methodology: (A) patient-specific gene network modeling, (B) computation of four classes of topological features, and (C) evaluation for patient classification, survival analysis, and therapeutic response prediction. © CEA
Our study highlights the utility of gene co-expression networks as an alternative to single-gene markers for improving the prediction of therapeutic response in cancer patients.
Fundings
This work was supported by
- the KATY and CANVAS projects under the European Union’s Horizon 2020 Research and Innovation programme
- the DIGPHAT project through the France 2030 initiative, within the PEPR Digital Health programme of the French National Research Agency (ANR).