Contexte Pour assurer la stabilité génomique et donc la protection contre le développement tumoral, les cellules ont établi un réseau complexe de voies métaboliques qui coordonnent le contrôle du cycle cellulaire, la réparation de l'ADN, l'apoptose et la sénescence. Ce réseau constitue la réponse aux dommages de l'ADN (DDR).Les cassures double brin (CDB) sont les lésions les plus toxiques. La recombinaison homologue et la religature d’extrémités non-homologues (NHEJ) constituent les principales voies de réparation des cassures double brin (DSBR). Depuis de nombreuses années, l'équipe s’intéresse à l'analyse des mécanismes de réparation des CDB. En utilisant des substrats intrachromosomiques, l'équipe a contribué à caractériser la voie alternative du NHEJ ( Guirouilh-Barbat et al, 2004, 2007), à identifier le rôle de Mre11 ( Rass et al, 2009) dans ce mécanisme et a également participé à identifier le rôle BLM de 53BP1 dans le contrôle de la réparation des CDB ( Grabarz et al, 2013).
Plus récemment, l'équipe se concentre sur la réparation de l’ADN et la sénescence en relation avec l'intégrité de la membrane nucléaire.
Il est proposé que la sénescence empêche la prolifération des cellules portant de l'ADN endommagé, constituant ainsi une barrière contre le développement tumoral. Cependant, la sénescence est une épée à double tranchant, car les données récentes ont proposé que les cellules sénescentes pourraient favoriser la prolifération tumorale en sécrétant des facteurs inflammatoires. Ainsi, les différentes voies de contrôle de la sénescence doivent être étroitement contrôlées et coordonnées. Depuis la théorie des radicaux libres du vieillissement proposé par Harman dans les années 1950, le stress oxydatif demeure l'une des causes les plus fréquemment citées pour le vieillissement. Toutefois, le contrôle moléculaire précis de la sénescence induite par le stress oxydant est loin d'être complètement élucidé. En plus du stress oxydant, l'érosion des télomères, des défauts dans la DDR et les modifications dans l'architecture nucléaire sont également associés à un vieillissement prématuré. L'interaction potentielle entre ces différents processus conduisant à la sénescence reste mal comprise, et aucun modèle unificateur ne pouvait être construit. Les syndromes progéroïdes ont souvent été classés en deux catégories: d’une part, les laminopathies, comme la progeria Hutchinson-Gilford (HGP), associées à des altérations de l’architecture nucléaire résultant de la dérégulation des lamine A/C, et d’autre part, les syndromes associés à des défauts de la DDR, comme l'ataxie télangiectasie (A-T). Les lamines A/C, B1 et B2 sont les principaux constituants de la lamina nucléaire, qui borde la membrane nucléaire interne et détermine sa forme et son intégrité. Sur la base de leur localisation à la périphérie du noyau, les lamines modulent l'expression des gènes, en interagissant avec la chromatine ou en séquestrant des facteurs de transcription. En outre, d'autres rôles pour les lamines dans le contrôle de la mitose, la réplication de l'ADN ou de la réponse aux dommages de l'ADN ont récemment émergé. | | Contribution récente Nous avons récemment découvert un nouveau mécanisme d’induction de la sénescence reliant le stress oxydatif à l’altération de l’architecture nucléaire.
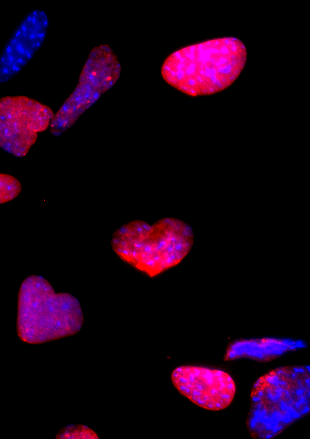
Nous avons montré une accumulation de la lamine B1 dans les cellules A-T et une déformation de l’enveloppe nucléaire. De façon importante, la surexpression de la lamine B1 est suffisante pour induire cette déformation et la sénescence dans des fibroblastes contrôles. De plus, la normalisation du niveau de la lamine B1 réduit simultanément les altérations de l’enveloppe nucléaire et la sénescence des cellules A-T. De plus, nous avons montré que l’accumulation de la lamine B1 est induite par le stress oxydant et l’activation de la MAPK-P38. Ces données mettent en lumière un rôle majeur de la lamine B1 dans l’induction de la sénescence des cellules A-T (Barascu et al., 2012). Notre étude suggère un modèle dans lequel la lamine B1 s’accumule transitoirement en réponse aux ROS (espèces réactives de l’oxygène) pour protéger les cellules contre le stress oxydant. Cependant, en cas de ROS persistants ou chroniques (comme dans le cas d’A-T), le niveau prolongé de lamine B1 aurait des conséquences délétères en affectant l’architecture de l’enveloppe nucléaire et en induisant la sénescence. Comme la dérégulation de la lamine A est associée à une persistance de dommages de l’ADN, il est tentant de spéculer que la surexpression de la lamine B1, en altérant l’architecture nucléaire, puisse aggraver les défauts de signalisation ou de réparation dans des pathologies associées à un stress oxydant élevé. (comme A-T).
|