Predicting the structure of proteins and their interaction modes is a real challenge for structural biologists. In addition to the gap between the number of proteins whose sequence is known and the number of available structures, proteomics has recently revealed the previously hidden part of the iceberg: hundreds of thousands of physical interactions between proteins. Knowing the surfaces involved in these interactions is essential, not only for understanding the mechanisms that govern how cells and organisms function, but also for designing new therapeutic or enzymatic molecules for pharmaceutical and biotechnological purposes.
Jessica Andreani and Raphaël Guérois (Team “Molecular Assemblies and Genome Integrity”/LBSR/I2BC) have been working for several years on the modelling of protein-protein interactions. In particular, they contribute to the improvement of prediction methods by integrating an evolutionary dimension into molecular docking tools. Indeed, protein interfaces tend to be more conserved than other regions on the protein surface. Moreover, signs of co-evolution can be detected at interfaces, where potentially disruptive mutations are compensated for by mutations in contacting positions on the protein partner. The team has thus developed and released the InterEvDock server, in collaboration with the Ressource Parisienne en Bioinformatique Structurale (RPBS, University of Paris). As in previous versions, the InterEvDock3 server offers a systematic search for possible interfaces between two partners (known as free docking) and generates numerous conformations that are then ranked, in particular by taking into account information on the evolution of protein sequences. This unique modelling server processes user requests in a variety of formats (structural or sequence data, on one or both partners).
The software is now in its third version (InterEvDock3). This version integrates three new prediction modes that are described in two articles published in
NAR1 and
Bioinformatics2. The first mode, which is not based on free docking, allows homology modelling of large complexes with potentially low sequence identity. It uses sequences (no structures) as input and runs a template-based modelling protocol by searching exhaustively for close and distant homologs to generate assembly models.
The second mode predicts the structure of complexes from contact maps resulting from methods combining covariation analysis and deep learning. This mode uses 3D structures of monomers or homomultimers (such as a helicase hexamer) to perform a free docking approach while trying to satisfy the contacts predicted in the contact map. It is able to handle some ambiguous information, especially if one of the two partners is a homomultimer (with its residues thus present several times in the structure) and a contact in the predicted map can thus materialise in different ways.
Finally, the third mode uses 3D structures of monomers or multimeric complexes (possibly modelled from sequences by mode 1) and implements a new strategy for evaluating interfaces with coevolutionary information. Ten to forty representative pairs of homologous sequences (i.e. ten to forty evolutionarily conserved interactions between homologs) are selected, modelled at the atomic scale and scored. This new algorithm, tested on a database of 752 complexes (see Bioinformatics2), increases the number of correctly predicted complexes by 30%.
It usually takes between 20 and 60 minutes for the server to propose an interaction model. Developed with funding from two national health and biology infrastructures (FRISBI and IFB), the server is accessible from the RPBS platform: https://bioserv.rpbs.univ-paris-diderot.fr/services/InterEvDock3/
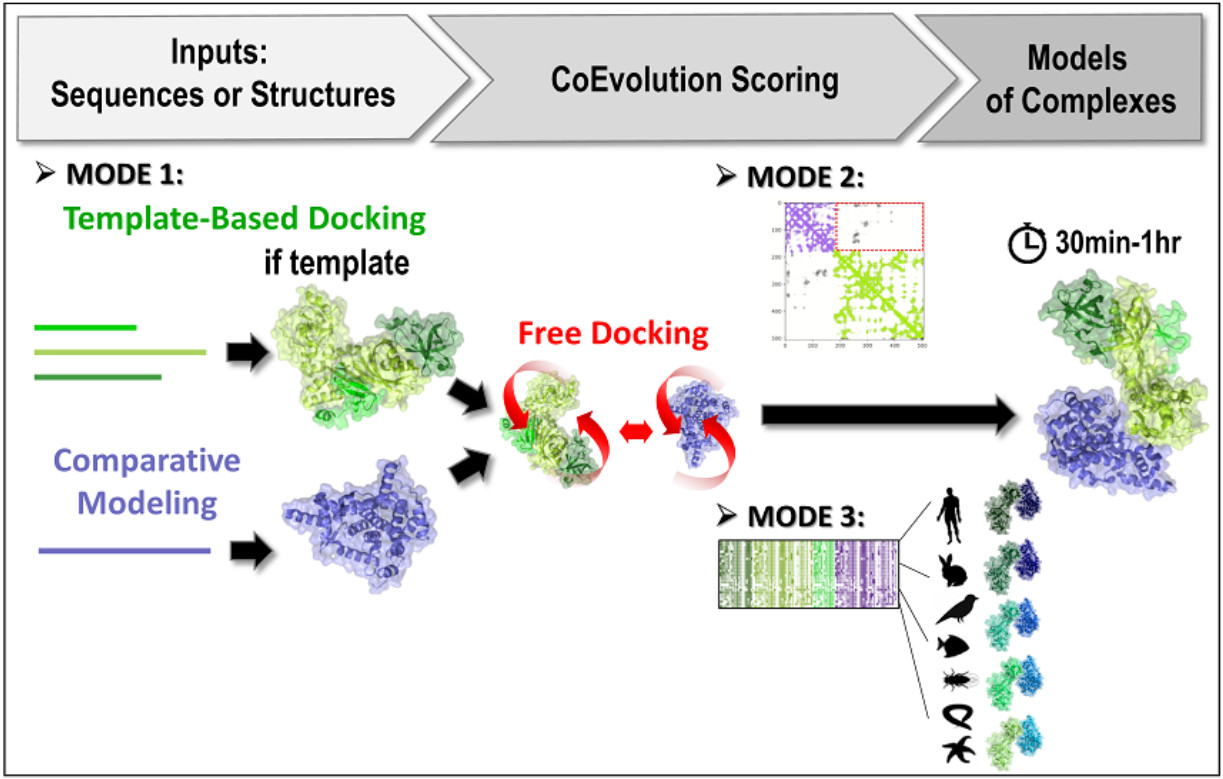
The 3 prediction modes of InterEvDock3.
Another server to analyze interaction data obtained by proteomicsThe team also collaborates with the RPBS on the Proteo3Dnet
3 server designed to analyze interactions identified by proteomic techniques by integrating structural information (in particular 3D structures of known complexes). Developed with the funding of three national infrastructures in health and biology (
FRISBI,
ProFI and IFB), it is accessible from the RPBS:
https://bioserv.rpbs.univ-paris-diderot.fr/services/Proteo3Dnet/