Beta thalassemia is a genetic disease affecting the red blood cells. It is characterized by a total (β⁰) or partial (β⁺) deficit in the synthesis of the β-globin subunit of hemoglobin. Patients with β-thalassemia major produce little or no β-globin and need regular blood transfusions to compensate for the deficit in blood cells and hemoglobin A, the most abundant form of the molecule, composed of two α-globin and two β-globin subunits. Researchers from IDMIT's Stem Cells and Therapeutic Applications Laboratory (LCSAT) developed a lentiviral gene therapy enabling the expression of the β-globin¹ gene. The clinical results they obtained between 2010 and 2019 led to a market authorization for the use of their therapy in patients with β⁺ thalassemia, i.e., those who have only a partial deficit in the subunit. Building upon those results, the researchers are continuing their efforts to improve the efficacy of the vector transfer of the gene, on one hand to expand the indication to patients with β⁰ disease (producing no β-globin) and on the other to reduce the number of vector copies per cell for all patients, and in so doing reduce the risk of insertional mutagenesis.
In patients with β-thalassemia, erythroid cell (red cell precursors) maturation is not blocked by the deficit in β-globin subunits per se, but by an excess of α-globin subunits, which are highly unstable and toxic when they are not associated with their β-globin partner. That aspect manifests in the fact that patients with β-thalassemia in whom one or two of the four copies of genes coding for α-globin (HBA1 and HBA2) are dysfunctional produce more functional red blood cells (with functional hemoglobin) than do those in whom all HBA1 and HBA2 copies are working.
To improve the efficacy of their lentiviral gene therapy, the researchers inserted, in one of the introns of the therapeutic β-globin gene, an additional gene sequence coding for an interfering RNA² targeted to HBA2. In their study published in Molecular Therapy, they showed that this new approach reduced the number of copies needed to restore α- and β-globin balance in patients with β-thalassemia nearly two-fold compared to the current market-approved treatment.
They furthermore verified that the novel lentiviral gene therapy was as stable as its predecessor, that it produced the same amount of therapeutic β-globin, and that the specific targeting of the RNA interference to HBA2 did not reduce the total quantity of α-globin subunits by more than 50%. Indeed, red blood cells remain perfectly functional even when one of the two genes coding for α-globin is dysfunctional.
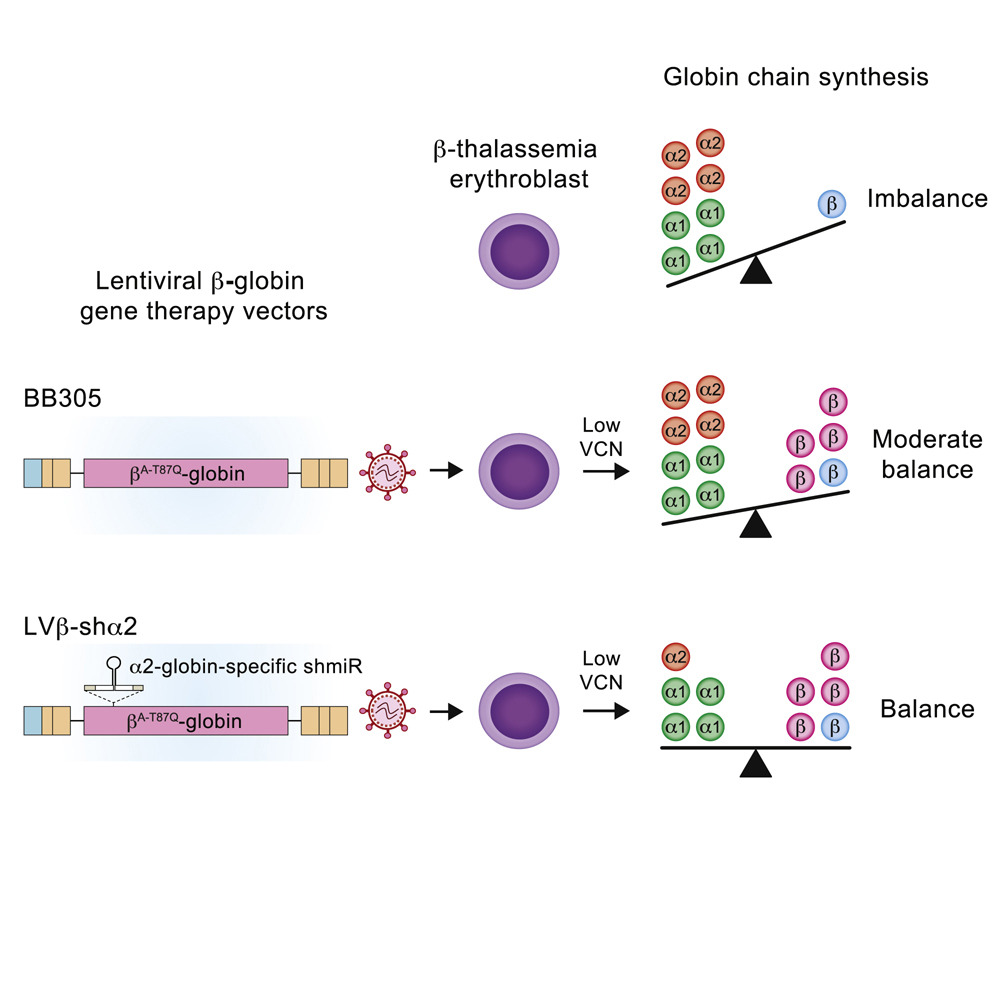
These very encouraging results suggest new therapeutic opportunities for patients with severe β⁰ thalassemia and improved benefit-risk ratios for all patients with β-thalassemia benefiting from this gene therapy.
This study was carried out in partnership with the Hudson Institute of Medical Research (Australia) and Mahidol University (Thailand).
1 : Institut de biologie François Jacob - Nouveau succès de thérapie génique dans la bêta-thalassémie (cea.fr) (french presse release)
2 : RNA interference is a process in which a single or double stranded RNA molecule diminishes the expression of a particular gene by interfering with the associated messenger RNA.