Prédire la structure des protéines et leurs modes d’interactions constitue un véritable défi pour les bio-informaticiens spécialistes de biologie structurale. Outre le fossé entre le nombre de protéines dont la séquence est connue et le nombre de structures disponibles, la protéomique a récemment révélé la partie jusque-là immergée de l’iceberg : des centaines de milliers d’interactions physiques entre protéines. Or, connaître les surfaces impliquées dans les interactions est essentiel, non seulement pour comprendre les mécanismes qui régissent le fonctionnement d’une cellule ou d’un organisme, mais aussi pour la conception de nouvelles molécules thérapeutiques ou enzymatiques pour la pharmacologie et les biotechnologies.
Jessica Andreani et Raphaël Guérois (équipe
Assemblages Moléculaires et Intégrité du Génome/LBSR/I2BC) s’intéressent depuis plusieurs années à la modélisation des interactions protéines-protéines. Ils contribuent notamment à l’amélioration des méthodes de prédiction en intégrant une dimension évolutive aux outils de « docking » moléculaire. En effet, les interfaces protéiques tendent à être plus conservées que les autres régions à la surface des protéines. De plus, des signes de co-évolution peuvent être détectés aux interfaces, où des mutations potentiellement perturbatrices sont compensées par des mutations dans des positions de contact sur le partenaire protéique. L’équipe a ainsi développé et mis à disposition, en collaboration avec la Ressource Parisienne en Bioinformatique Structurale (RPBS, Université de Paris), le serveur InterEvDock. Comme dans les versions précédentes, ce serveur propose une recherche systématique des interfaces possibles entre deux partenaires et génère de nombreuses conformations (on parle de « docking » libre) qu’il classe notamment en tenant compte d’informations sur l’évolution des séquences protéiques. Ce serveur de modélisation unique traite des requêtes d’utilisateurs de format varié (données structurales ou seulement séquences, données sur l’un des partenaires ou les 2).
Le logiciel en est désormais à sa troisième version (InterEvDock3). Celle-ci intègre 3 nouveaux modes de prédiction qui sont décrits dans deux articles publiés dans NAR1 et Bioinformatics2. Le premier mode, qui n’est pas basé sur du docking libre, permet de modéliser par homologie de gros complexes à basse identité de séquence. Il utilise en entrée des séquences (aucune structure) et exécute un protocole de modélisation basé sur des modèles en recherchant des homologues proches et éloignés de manière exhaustive pour générer des modèles d'assemblages.
Le deuxième mode permet de prédire la structure de complexes à partir des cartes de contacts résultant de méthodes combinant des analyses de covariation et d’apprentissage profond. Ce mode utilise des structures 3D de monomères ou d’homomultimères (comme un hexamère d’hélicase) pour exécuter une approche de docking libre en essayant de satisfaire les contacts prédits dans la carte de contacts. Il est capable de traiter certaines informations ambiguës, en particulier si l’un des deux partenaires est un homomultimère (avec donc ses résidus présents plusieurs fois dans la structure) et qu’un contact de la carte prédite peut donc se matérialiser de différentes façons.
Enfin, le troisième mode utilise des structures 3D de monomères ou de complexes multimères (éventuellement modélisés à partir des séquences du mode 1) et met en œuvre une nouvelle stratégie d’évaluation des interfaces avec des informations sur la coévolution. De 10 à 40 paires représentatives de séquences homologues (c’est-à-dire 10 à 40 interactions conservées entre homologues au cours de l’évolution) sont sélectionnées et modélisées à l’échelle atomique et un score leur est attribué. Ce nouvel algorithme, testé sur une base de données de 752 complexes (voir Bioinformatics2), permet d’augmenter de 30% le nombre de complexes correctement prédits.
A titre indicatif, il faut généralement entre 20 et 60 minutes au serveur pour proposer un modèle d’interaction. Développé avec le financement de deux infrastructures nationales en biologie santé (FRISBI et
IFB), le serveur est accessible depuis la RPBS :
https://bioserv.rpbs.univ-paris-diderot.fr/services/InterEvDock3/
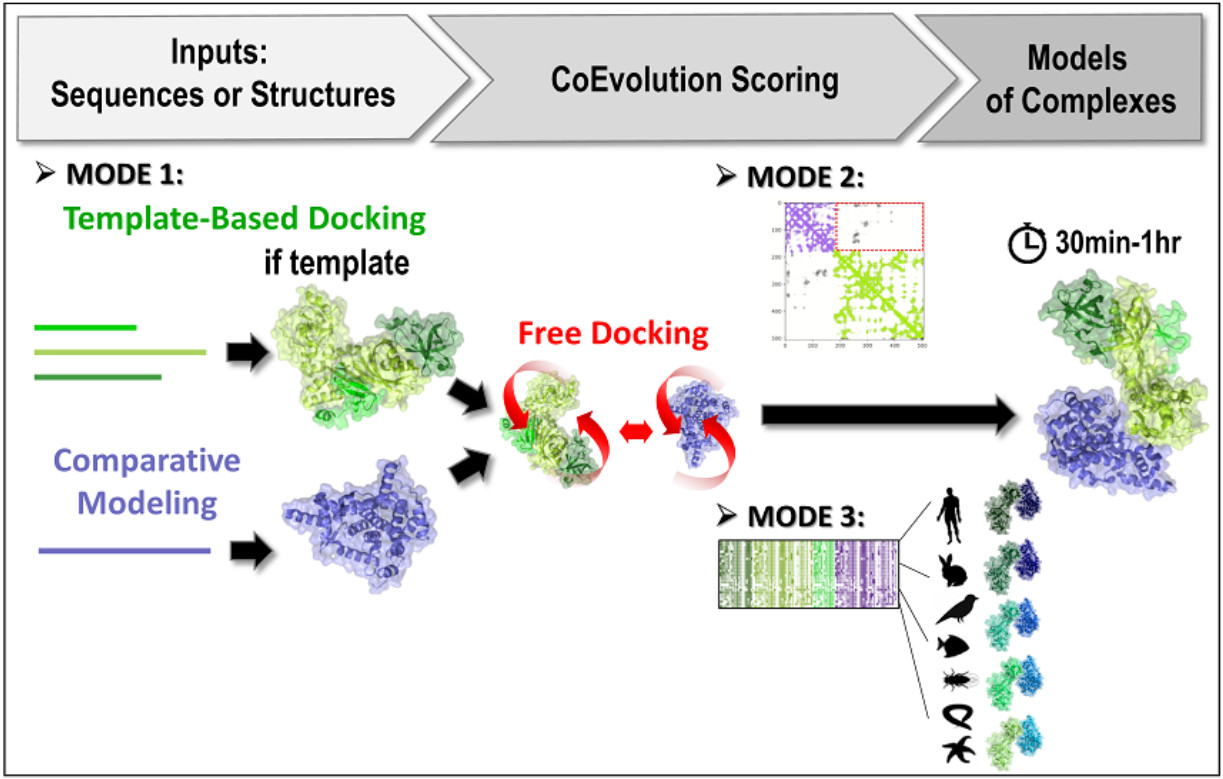
Représentation schématique des 3 modes de fonctionnement d'InterEvDock3.
Un autre serveur pour analyser les données d'interactions obtenues par protéomiqueL'équipe collabore également avec la RPBS sur le serveur Proteo3Dnet
3 conçu pour analyser les interactions identifiées par des techniques de protéomique en intégrant des informations structurales (notamment des structures 3D de complexes connues). Développé avec le financement de trois infrastructures nationales en biologie santé (
FRISBI,
ProFI et IFB), il est accessible depuis la RPBS :
https://bioserv.rpbs.univ-paris-diderot.fr/services/Proteo3Dnet/
Contacts Joliot :
Jessica Andreani (jessica.andreani@cea.fr)
Raphaël Guérois (raphael.guerois@cea.fr)